El déficit de alfa-1-antitripsina (α1-AT) es una enfermedad congénita causante de hepatopatías en niños y enfisema pulmonar en adultos, siendo en éstos una condición potencialmente mortal. A pesar de ello, es una enfermedad subdiagnosticada y en los adultos se suele llegar al diagnóstico en etapas muy avanzadas de la enfermedad pulmonar. La Organización Mundial de la Salud recomienda realizar la cuantificación sérica de α1-AT a todos los pacientes con EPOC y a los niños con trastornos hepáticos en la primera infancia.
La α1-AT es una glicoproteína producida en los hepatocitos, cuya función principal es inhibir las proteasas neutrofílicas, especialmente la elastasa leucocitaria. Además, se le atribuyen propiedades antiinflamatorias, antimicrobianas e inmunomoduladoras. Sus niveles pueden aumentar en situaciones de inflamación o daño tisular, comportándose como un reactante de fase aguda.
Los pacientes con déficit de α1-AT pueden tener afección hepática y/o pulmonar. La sustitución de un aminoácido resulta en una proteína anormal que no puede ser secretada al plasma, acumulándose en el retículo endoplásmico del hepatocito y causando lesión hepática. El daño pulmonar se da como consecuencia de la destrucción alveolar producida por la actividad proteolítica de la elastasa de los neutrófilos sobre el tejido conectivo; no inhibida debido a los bajos niveles de α1-AT en suero y líquido pulmonar. El daño hepático aparece solo en aquellas variantes con polimerización anormal y acumulo intrahepatocitario. Por otro lado, la aparición de enfisema se relaciona a las variantes deficientes o nulas.
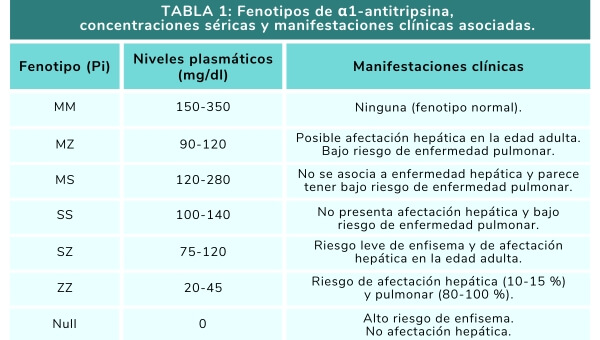
Las variantes fenotípicas se nombran en función de su movilidad electroforética como Pi (inhibidor de proteasas) seguido de unas letras mayúsculas que corresponden a los alelos heredados de ambos progenitores (su herencia es codominante) (Tabla 1).
- La variante M es la más frecuente y se asocia con niveles séricos y actividad normal.
- Las variantes Z y S se atribuyen a bajos niveles de α1-AT. La Z está vinculada con enfermedad hepática y pulmonar. La S no produce acúmulo intrahepatocitario significativo, por lo que no se asocia a enfermedad hepática.
- En las variantes nulas hay un error en la transcripción o traslación con interrupción de la síntesis proteica, cursando con niveles indetectables de α1-AT. Esta variante se relaciona con enfermedad pulmonar.
Clínica:
Enfermedad hepática:
Es la forma de presentación predominante en edad pediátrica. Es la causa del 10 a 15 % de las colestasis y hepatitis durante el periodo neonatal. Estos pacientes pueden presentar bajo peso, hepatomegalia, hiperbilirrubinemia conjugada, elevación leve a moderada de las transaminasas, fosfatasa alcalina y gamma-glutamil-transferasa e hipercolesterolemia. En algunos casos puede aparecer coagulopatía por déficit de vitamina K. Normalmente, la ictericia desaparece luego de las 2 a 4 semanas; pero un pequeño porcentaje puede desarrollar hepatopatía crónica y cirrosis. Un 16 % de los niños que presentan el fenotipo PiZZ evolucionan a insuficiencia hepática terminal, necesitando trasplante hepático antes de los 5 años de edad.
En los adultos, la afectación hepática es infrecuente y se puede presentar como hepatitis crónica, cirrosis o hepatocarcinoma. Cabe aclarar que no siempre existe el antecedente de enfermedad hepática neonatal.
Enfermedad pulmonar:
La prevalencia se estima entre 75 y 85 % de los pacientes con déficit grave de α1-AT (fenotipo PiZZ), apareciendo en la edad adulta a partir de los 30 años. Se manifiesta con disnea progresiva, tos y agudizaciones frecuentes y prolongadas. En muchos casos es difícil diferenciar estos síntomas de una EPOC. La enfermedad es de progresión lenta pudiendo acelerarse por infecciones repetitivas de las vías bajas, inhalación de polvo y otros irritantes como el tabaco.
Diagnóstico:
El déficit de α1-AT debe sospecharse en los casos de ictericia neonatal y en cualquier paciente con cirrosis hepática de etiología desconocida, independientemente de la edad; en especial si hay una historia previa de ictericia o enfermedad hepática durante la infancia. También hay que considerarlo en las personas con enfisema o EPOC, sobre todo si se desarrolla de forma temprana, sean fumadores o no.
Además, siempre debe realizarse el estudio de los familiares consanguíneos de los pacientes ya conocidos, aunque estén asintomáticos.
Para confirmar el diagnóstico tras la sospecha clínica, se debe determinar el valor de α1-AT en plasma. Si los valores son bajos, es preciso identificar el fenotipo mediante isoelectroenfoque. Cuando el fenotipo no se corresponda con los niveles esperados, habrá que estudiar el genotipo por PCR.
Generalmente se determinan los alelos deficitarios más frecuentes (S y Z), y en caso de no detectarlos se debe realizar la secuenciación completa del gen, para investigar la presencia de alelos raros.