En las últimas dos décadas, la Reacción en Cadena de la Polimerasa (PCR) se ha convertido en una herramienta indispensable para diagnóstico molecular, investigación biomédica y control de calidad industrial. La PCR en tiempo real (qPCR) marcó un salto importante al permitir la cuantificación de ácidos nucleicos, de manera más rápida y precisa que la PCR convencional en gel.
Sin embargo, una nueva generación tecnológica está ganando terreno: la PCR digital (dPCR). Esta metodología, aunque más reciente y todavía en proceso de adopción masiva, promete mejorar notablemente la sensibilidad, precisión y reproducibilidad de la cuantificación molecular.
La reacción en cadena de la polimerasa es un método enzimático in vitro para amplificar secuencias específicas de ADN o ARN. Se basa en el uso de la capacidad de la enzima ADN polimerasa para sintetizar una nueva cadena de ADN, complementaria a la cadena utilizada como molde o de interés.
Hay 6 componentes básicos que son similares entre todos los tipos de PCR:
1. Templado ADN / ARN: contiene la secuencia a amplificar.
2. Primers, cebadores o iniciadores: son oligonucleótidos que definen la secuencia a amplificar.
3. Desoxinucleótidos trifosfatos (dNTP): dATP, dTTP, dCTP, dGTP
4. ADN polimerasa termoestable
5. Cofactor de la enzima: por lo general son Iones Mg2+
6. Solución buffer: mantiene el pH y la fuerza iónica de la solución de reacción para el correcto funcionamiento de la enzima.
Una PCR se puede dividir en 3 grandes pasos:
El primero es una desnaturalización inicial, donde el ADN bicatenario se separa en dos hebras simples a una temperatura mayor a 90°C por un tiempo aproximado de 3 a 5 minutos; lo que va a depender del tamaño de nuestro ADN molde y el contenido de G/C (guanina/citosina).
El segundo paso, a su vez, se subdivide en 3 simples partes, llamado “ciclo de reacción” que se repite aproximadamente unas 40 veces. Aquí ocurre una desnaturalización en la que un calentamiento breve (30 segundos a 94°C) separa las hebras, un alineamiento o “Annealing” que permite a los cebadores formar enlaces de hidrógeno con los extremos de la secuencia objetivo, y una extensión donde la ADN polimerasa adhiere nucleótidos en el extremo 3´ de cada cebador, permitiendo que las moléculas de ADN se dupliquen en cada ciclo de reacción.
El último paso es una extensión final, por lo general a 72° de unos 5 a 10 minutos que como resultado nos dará nuestro fragmento final de interés.
El gráfico típico de amplificación por PCR en tiempo real muestra una curva con forma sigmoidea: incluye una fase basal, seguida de una fase exponencial que alcanza una meseta a través de una fase lineal. La fase exponencial representa el estadío más eficiente de amplificación y la cantidad de productos de PCR se duplica con cada ciclo si la eficiencia de amplificación es del 100 %. El método es cuantitativo debido a que se calibra con una curva estándar. La cantidad “absoluta” de secuencia objetivo en una reacción de PCR se mide en relación con una curva estándar, generada a partir de una muestra de cantidad conocida o número de copias.
Por otra parte, dPCR es una técnica que permite medir de forma exacta la cantidad de ADN o ARN objetivo en una muestra, superando algunas limitaciones con respecto a otras técnicas de PCR.
Sus principios son:
1. Preparación del templado ADN / ARN
2. Partición o división
3. Amplificación
4. Detección y análisis de resultados.
En dPCR la muestra se divide en muchas reacciones pequeñas e independientes dentro de una placa de reacción. Cada una de estas “particiones” puede contener unas pocas moléculas objetivo o ninguna.
El siguiente paso es la amplificación: además de nuestra secuencia de interés, en cada pocillo se encuentra toda la maquinaria necesaria para que nuestra sonda hibride y sea hidrolizada por una enzima, la Taq Polimerasa; de esta forma libera el fluoróforo de nuestro Quencher y así puede emitir fluorescencia. Luego, se calcula cuántas particiones dieron resultado positivo.
Con esos datos y aplicando un cálculo estadístico de Poisson, se determina la concentración exacta del material genético.
Dividir la muestra de esa forma nos permite concentrar mejor las moléculas de interés, lo que reduce la competencia entre ellas y facilita la detección de mutaciones muy poco frecuentes dentro de un gran fondo de secuencias normales. Además, esta técnica es más tolerante a sustancias inhibidoras que puedan estar presentes en el material analizado.
Para resumir, podemos observar las diferencias más importantes entre dPCR y una qPCR:
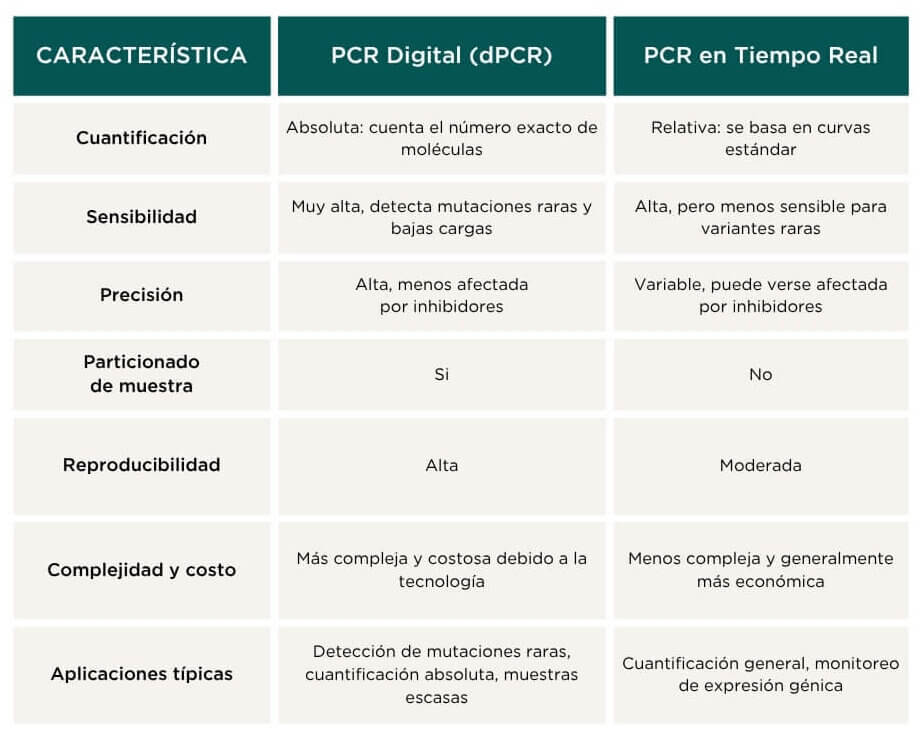
Como conclusión, la PCR digital representa una evolución significativa frente a la PCR en tiempo real, posicionándose como una herramienta clave para el futuro del diagnóstico molecular. Su capacidad para realizar una cuantificación absoluta sin necesidad de curvas estándar, junto con su mayor sensibilidad y precisión en la detección de variantes genéticas o bajas cargas virales, la convierte en una opción superior. Además, su tolerancia a inhibidores y su robustez en muestras complejas permiten resultados más confiables y reproducibles. A medida que los costos disminuyen y la tecnología se vuelve más accesible, se prevé que la PCR digital pueda reemplazar progresivamente a la PCR en tiempo real en múltiples aplicaciones clínicas, ambientales y de investigación, consolidándose como el nuevo estándar en análisis genético de alta resolución.